CompGen is a research consortium co-led by Prof. Iyer, Prof. Lumetta, and Prof. Robinson, comprising ECE, CS, and IGB researchers and students with a goal of building a machine to accelerate computational genomics (example: variant calling, phylogeny). Collaborations include researchers from Mayo Clinic, IBM, Intel, Monsanto, Eli Lilly, Strand Life Sciences, etc. to make this a practical deployment with the future of individualized medicine and genomics-as-service being imminent.
Research Staff:
- Subho Sankar Banerjee
- Mohamed El Hadedy Aly
- Zachary Stephens
- Arjun Athreya
- Safa Messaoud
- Ching Yang Tan
Projects
Next-Generation Sequencing Simulator
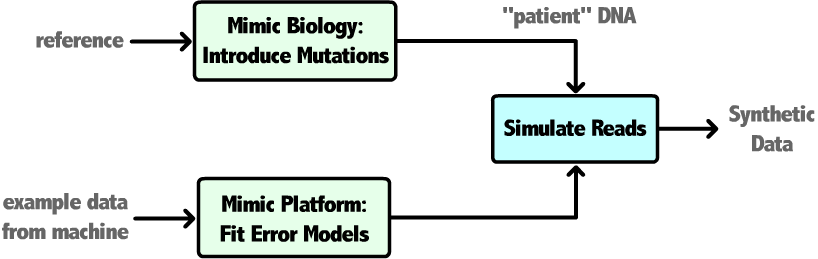
This project has focused on developing deeper understanding of accuracy throughout next-generation sequencing experiments. Synthetic datasets and specialized analysis pipelines have been created to quantify the relationship between input dataset characteristics and errors introduced by each step of a variant-calling workflow. This work allows us to provide bioinformaticians a priori estimates of alignment and variant-calling accuracy for their pipeline, useful for software benchmarking and workflow design. Additionally, we will incorporate limitations in alignment accuracy imposed by the inherent repetitiveness present in the DNA of most organisms. Ongoing work will combine these efforts into a module that can provide CompGen users with a selection of candidate pipelines based on approximated attributes of their data that offer performance/accuracy tradeoffs.
Computational Kernels and Acceleration of Genomics Tools
We extracted the computational kernels that are at the heart of several genomic analyses, which could be potentially implemented in hardware. Core computational kernels were extracted by studying over 40 tools encompassing popular genomic analyses such as phylogeny, sequence alignment, variant calling, and metagenomics. This study shows that there exist only 4 major computational kernels (linear algebraic product sums for clustering, hidden Markov models with Viterbi algorithm, DeBruijn graph construction, and maximum weight trace algorithm in dynamic programming problems) that encompass several genomic analyses. In particular, we have studied the most popular variant-calling tool GATK Haplotype Caller. Our initial finding is that programming inefficiencies constitute 70% of run time and core computation kernels such as hidden Markov modeling (HMM) and the dynamic programming problem of Smith-Waterman algorithm comprise the remaining 15%. This collaboration has also studied the possible ways of overcoming the computing time due to programming efficiencies, which alone gives 60–70% speedup in software. Based on current implementations of HMM and Smith-Waterman on accelerator hardware such as graphics processing units (GPU) or field-programmable gate arrays (FPGA), and with overcoming programming inefficiencies, our initial calculations show that for chromosome 13, the variant-discovery time is decreased from 20 minutes to about 5 minutes. The next step in the study is to unearth the computations of these analyses, which could be better run on accelerators such as GPUs by exploiting data parallelism versus those better run as single-threaded processes on commodity central processing units (CPUs). This will highlight the need of technologies such as IBM’s CAPI, a new hardware framework to integrate heterogeneous accelerators.
Computational Genomics Framework
We have studied the execution traces (performance profiles) of various genomic analyses/algorithms on a variety of computation platforms, ranging from commodity CPU, GPU, and GPU of the Blue Waters supercomputer. These profiles (which include CPU hardware performance counters, memory hierarchy usage, and disk and network IO usage) give a holistic view of how the system performs when processing chromosomes, exomes, and whole genomes. Our results, in collaboration with HPCBio and NCSA researchers (L. Mainzer and V. Kindratenko), show that even though these genomic analyses are input-output (IO)-intensive computations, they are not particularly IO limited on machines like Blue Waters; this is contrary to both popular opinion and results published for older Beowulf-like clusters. Additionally, we are proposing the framework combining the study of computational kernels and the measurements from Blue Waters.